如何在细胞群中找到标记基因
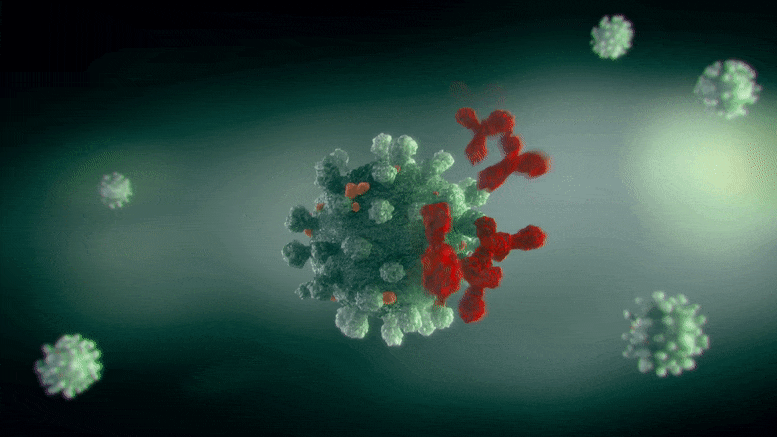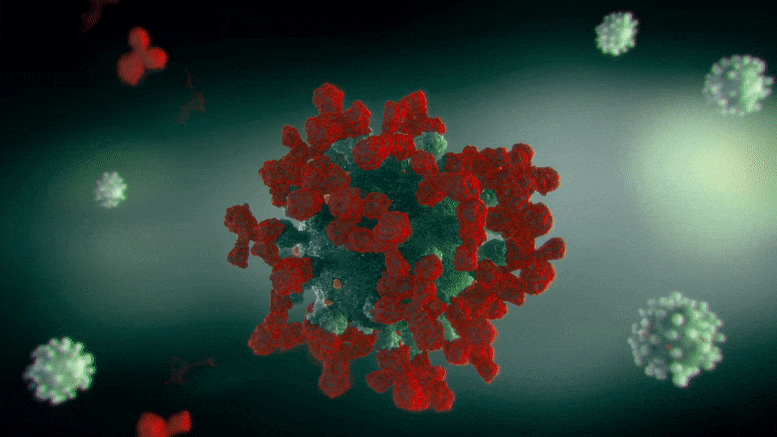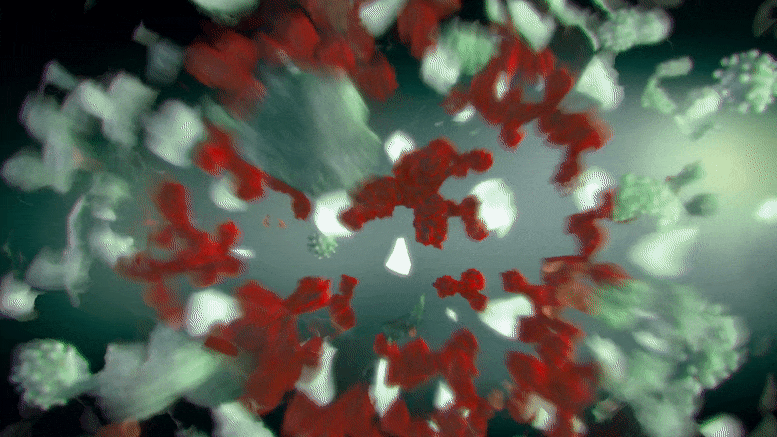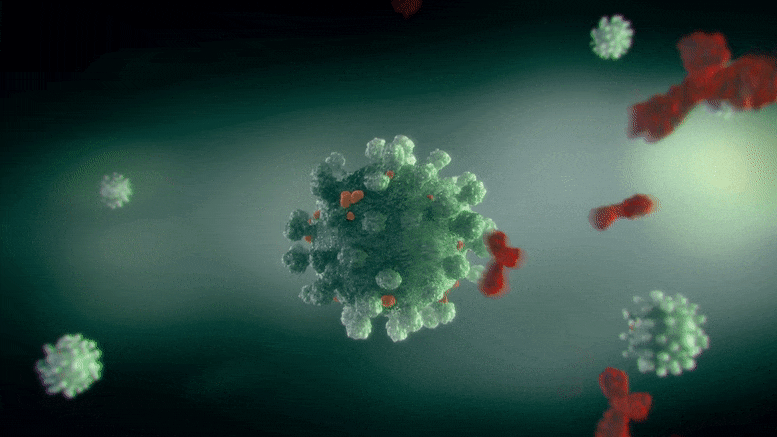
ApL包生成的细胞群关联图,基因用点表示。右边的颜色刻度代表基因集群关联的强度。红色的基因是最具集群特异性的基因。
一个生物样本中成千上万个细胞各不相同,可以一个细胞一个细胞地分析。根据它们的基因活动,它们可以被分类成簇。但是,哪些基因是一个特定集群的特别特征,即什么是它的“标记基因”?一种新的统计方法称为关联图有助于确定和分析这些标记基因。
哪些基因是特定的细胞类型,即“标记”他们的身份?随着当今数据集规模的不断扩大,回答这个问题往往是具有挑战性的。通常,标记基因只是在特定细胞群中发现的基因。然而,还有更多的基因可能是特定细胞类型的特征,但仍未被发现。
“关联图(ApL)”是一种新的统计方法,用于可视化细胞集群内的基因活动,使其更容易找到标记基因。这些图比较了一个给定集群的基因活动与数据集中所有其他集群。此外,它们可以很容易地看到哪些基因与其他集群共享.
“关联图不仅允许我们识别新的标记基因。它也可以反过来工作——我们能够根据提供的标记基因列表,将数据集中身份未知的集群与细胞类型相匹配,”柏林马克斯·普朗克分子遗传学研究所的Elzbieta Gralinska说。
这位生物技术专家所在的Martin Vingron团队开发了这项技术,并在两个公开的数据集上演示了它的功能,并公布了结果。此外,ApL已经作为统计环境的免费模块发布。ApL程序包使研究人员能够直观地检查他们的单细胞数据,并通过光标选择单个基因来了解更深入的细节。
分析和分组单个细胞为什么有必要首先识别标记基因?现代测序技术能够破译单个细胞中的单个RNA分子。例如,从血液样本中,每个细胞都可以被分离出来,细胞的RNA样本也可以被解码。这些单细胞数据代表转录成RNA分子的活性基因。
它的优点是:不用纠结于特定RNA属于哪种细胞类型,它可以追溯到它的细胞起源。缺点是:在数以万计的细胞中,对每一个细胞中的数千个RNA进行测序,会产生大量的数据。
一种方法是根据RNA含量对细胞进行分类。单细胞数据是由许多不同细胞类型混合而成的。我们感兴趣的是相同细胞类型的细胞,它们的行为应该是相似的,”Martin Vingron解释说。因此,通过计算将相似的细胞分组是有意义的,他说。“对我们来说,标记基因定义了细胞类型。”
交互式地探索细胞群利用公开的白细胞数据,该团队演示了新算法的工作原理。许多不同类型的白细胞,如T细胞、B细胞或单核细胞,都被分成不同的簇。研究人员证实了已知的标记基因,并能够表明血细胞之间的近亲在基因活性上也有很大的相似性。
“我们用ApL发现的每一个标记基因可能已经被至少一种其他现有的标记基因鉴定方法所发现,”Gralinska说。但与现有算法相比,ApL的优势在于其结果的图形表示,她说。“现有的工具提供了长长的基因列表和得分值。通常,用户浏览列表时,会在一个任意的分界点停下来。”Gralinska说。
她说,相比之下,新方法提供了一种可视化这些基因的方法,点击每个基因并仔细观察其活动。“我们不仅提供了标记基因的列表,我们还允许用户回顾这些基因的行为,”研究人员说。“有了关联图,他们可以深入研究数据,更多地了解每种细胞类型。”此外,她说,通过基因本体论术语富集分析,在后续步骤中很容易分解最有趣的基因的生物学作用,这与ApL软件兼容——她认为这是“一个非常有用的功能”。
潜在的数学模型包含跨基因活动信息的高维数据不能在不丢失信息的情况下以视觉方式表示。对于集群数据也是如此,所有这些都使分析变得复杂。Gralinska说:“我们的技巧在于,我们考虑的不仅仅是二维或三维,而是最终创造出一个二维图表。”
关联图来自于一种数学技术,同时嵌入基因和细胞在一个共同的,高维空间。在这个空间中,测量基因和给定的细胞集群之间的距离会产生成对的值,这些值反映了一个基因与给定的集群的关联,并对其与其他集群的关联提供了见解。
“ApL的一个缺点是,我们依赖于聚类前的数据,这意味着我们必须依赖其他技术来聚类,”Martin Vingron说。“尽管如此,我们希望我们的新方法能找到许多新用户。我们发现,视觉和互动过程可以更好地进行分析。”
Visualizing Cluster-specific Genes from Single-cell Transcriptomics Data Using Association plots