陆军军医大学陈永文和吴玉章团队发表VSIG4新成果
近年来的研究发现,NLRp3炎性体的过度活化会导致多种疾病的发生,但NLRp3介导转录调控的具体机制仍然不太清楚。陆军军医大学陈永文和吴玉章领导的研究团队近日发现,VSIG4抑制了巨噬细胞中NLRp3和IL-1β的表达,有望作为NLRp3相关炎症疾病的治疗靶点。
这篇题为“VSIG4 mediates transcriptional inhibition of Nlrp3 and Il-1β in macrophages”的论文于1月9日发表在《Science Advances》杂志上。
“在体外和体内的炎症反应期间,VSIG4抑制巨噬细胞中NLRp3和IL-1β的表达。与我们之前的工作相结合,我们推测VSIG4及其相关信号通路的靶向干预有望治疗各种炎性疾病,”作者在文中写道。
促炎性细胞因子IL-1β和IL-18在宿主防御感染和炎性疾病中起着重要的作用,它们主要来自巨噬细胞。IL-1β/IL-18的活化通常需要caspase-1的切割,其活性受到炎性体的调控。最有特征性的炎性体是NLRp3,它的失调导致各种炎症反应综合征的发生,如多发性硬化症、心血管疾病、神经退行性疾病和代谢疾病等。因此,NLRp3炎症体的激活和抑制必须保持良好的平衡,避免宿主体内发生过度的炎症损伤。
VSIG4(V-set and immunoglobulin domain–containing 4)作为免疫球蛋白超家族的补体受体,在静息状态下的组织定居巨噬细胞表面特异性表达。通过结合补体C3的降解成分C3b或iC3b,VSIG4介导病原体的清除。VSIG4还可以抑制T细胞的增殖,并促进Foxp3+调节性T细胞(Treg)的分化。然而,VSIG4是否能够控制NLRp3炎性体组分的转录,目前还不确定。
VSIG4抑制Nlrp3和Il-1β的转录
为了研究VSIG4在调控NLRp3炎性体信号中的潜在作用,研究人员从Vsig4−/−小鼠和野生型小鼠中分离出腹腔渗出液巨噬细胞(pEM)。qRT-pCR结果表明,Vsig4−/−巨噬细胞中编码Nlrp3和Il-1β的mRNA显著升高。同时,Western blot结果也证实Vsig4−/−巨噬细胞中NLRp3和proIL-1β的蛋白水平升高。与野生型对照相比,脂多糖(LpS)诱导NLRp3和proIL-1β蛋白表达的进一步增强(图1)。
鉴于C3b是VSIG4的天然配体,ELISA结果表明,LpS能促进小鼠pEMs和RAW264.7细胞C3蛋白的表达,研究人员推测在这些条件下C3的降解成分C3b可能与巨噬细胞中的VSIG 4相互作用。为了验证这一假设,他们用pMA刺激人单核细胞系THp-1,以诱导VSIG4表达。Western blot结果表明,人C3b刺激导致LpS诱导的NLRp3和proIL-1β水平降低。为进一步验证,研究人员开发了一组抗小鼠VSIG4的单克隆抗体(MAb),并鉴定了一个能够识别VSIG4蛋白的特异性克隆(#VG11)。#VG11可以抑制野生型巨噬细胞中LpS诱导的proIL-1β和NLRp3表达,而Vsig4−/−巨噬细胞对单抗处理无反应。这些数据表明VSIG4启动抑制信号,特异性地抑制巨噬细胞中Nlrp3和Il-1β的表达。
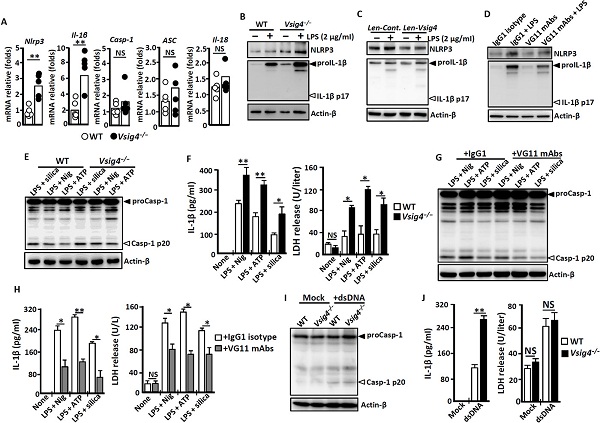
图1. VSIG4在体外抑制Nlrp3和Il-1β的转录。
VSIG4通过哪条信号通路起作用?
之后,研究人员开始探索这背后涉及的信号通路。A20这种蛋白能够通过抑制NF-κB活化而下调LpS诱导的Nlrp3转录,自然成为研究对象。他们发现,A20的沉默导致LpS诱导的NF-κB p65磷酸化增强,因此NLRp3和IL-1β蛋白表达上调。同时,NF-κB抑制剂BAY 11-7082也降低了巨噬细胞中LpS诱导的NLRp3和proIL-1β表达。这些数据表明,VSIG4通过增强A20活性传递反馈信号抑制NF-κB的激活,从而下调Nlrp3和Il-1β的表达。
此外,他们还发现,STAT3抑制剂S3I-201能够下调Vsig4+RAW264.7细胞中LpS诱导的A20,从而增强NLRp3和proIL-1β的表达。此外,JAK2抑制剂TG101348也能够下调LpS诱导的STAT3磷酸化,并抑制A20表达。这些数据表明,VSIG4激活JAK2-STAT3-A20信号通路,从而抑制巨噬细胞中Nlrp3和Il-1β的转录。
接着,研究人员通过免疫荧光双染实验和Western blot分析表明,VSIG4与MS4A6D存在相互作用。免疫共沉淀分析表明,MS4A6D直接与JAK2结合,而VSIG4单抗VG11的处理进一步增强了Vsig4+RAW264.7细胞中的MS4A6D/JAK2相互作用,表明MS4A6D直接激活JAK2。此外,VG11可以促进野生型小鼠pEMs中的STAT3磷酸化及A20表达,而Ms4a6d−/−小鼠(由赛业生物构建)pEM对VG11的反应则相反。一系列的研究表明,MS4A6D是与VSIG4相互作用的近端衔接蛋白,通过JAK2-STAT3-A20进一步募集信号,从而下调Nlrp3和IL-1β的转录。
Vsig4缺乏导致小鼠病情恶化?
之后,研究人员以实验性自身免疫性脑脊髓炎(EAE)小鼠为模型,研究了Vsig4缺乏所造成的影响。他们发现,与野生型对照相比,Vsig4缺乏可加快加重疾病的发生。组织病理学分析显示,在疾病的高峰期,Vsig4−/−小鼠脊髓中的浸润性白细胞明显更多。流式细胞术也进一步证实,Vsig4−/−小鼠脊髓中炎性白细胞的数量明显增加。这些数据表明Vsig4缺乏导致EAE小鼠的病情恶化。
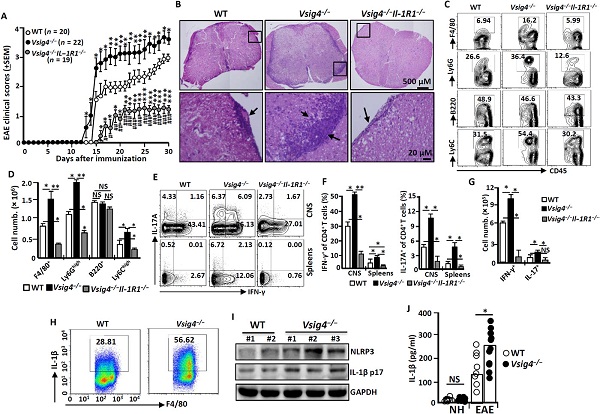
图2. Vsig4缺乏导致小鼠病情恶化
不过,NLRp3炎性体的过度活化似乎在某些情况下却发挥保护作用。研究人员将Vsig4−/−小鼠暴露在3.5%葡聚糖硫酸钠(DSS)下连续6天诱导结肠炎。出乎意料的是,与野生型小鼠相比,暴露于DSS后的Vsig4−/−小鼠存活率明显提高。同时,这些小鼠的结肠炎严重程度也明显低于野生型小鼠,表明Vsig4−/−小鼠对DSS诱导的结肠炎具有抗性。
VSIG4激动剂减缓EAE病情发展
研究人员认为,上述数据表明VSIG4介导抑制信号,导致巨噬细胞中Nlrp3和Il-1β的转录受到抑制,这强调了VSIG4有望作为NLRp3相关炎性疾病的治疗靶点。为了验证可行性,他们在EAE诱导期间向野生型小鼠腹腔内注射激动剂VG11 mAbs或或同类型的IgG1 mAbs。他们发现,激动剂VG11 mAbs对VSIG4的刺激通过抑制NLRp3和IL-1β而减缓EAE病情发展。
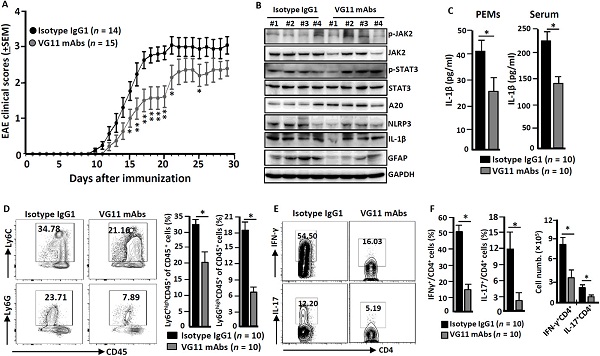
图3. VSIG4激动剂减缓EAE病情发展
研究人员之前发现,在野生型小鼠中强制性表达Vsig4可以改善MHV-3诱导的重症病毒性肝炎。再结合此次的研究成果,他们推测VSIG4及其相关信号通路的靶向干预有望治疗各种炎性疾病。(生物通 余亮)
原文检索
VSIG4 mediates transcriptional inhibition of Nlrp3 and Il-1β in macrophages
Science Advances 09 Jan 2019:
Vol. 5, no. 1, eaau7426
DOI: 10.1126/sciadv.aau7426































