董晨教授连发Nature,Cell子刊两篇文章解析肿瘤免疫治疗新发现
近年来, 肿瘤免疫治疗蓬勃发展, 已经给人类彻底战胜癌症带来了一线曙光。CD8+ T细胞是免疫系统攻击癌症细胞的主要利器。但是在肿瘤微环境里,CD8+ T细胞经常会进入机能缺陷或衰竭状态,从而不能有效地阻止癌症的进展。肿瘤细胞表面上调的抑制性配体pD-L1(programmed Death-Ligand 1)与CD8+ T细胞上pD-1(programmed Death-1)的相互作用是介导T细胞衰竭的机制之一;阻断pD-1/pD-L1通路的药物在癌症病人中疗效优于传统治疗手段,并已获FDA批准用于多种癌症的治疗。
然而,在大部分癌症类型中,只有小部分病人(20~30%)对anti-pD-1/pD-L1免疫治疗产生应答,非应答者一般经历体内T细胞功能短暂激活后疾病继续进展。因此, 肿瘤微环境pD-1可能与其他分子一起协同地促进T细胞衰竭。
B7 superfamily member 1(B7S1),也称B7-H4、B7x或VTCN1,在2003年被董晨、陈列平和James p. Allison三个实验室几乎同时发现为T细胞的共抑制分子。B7S1过度表达于多种癌症,因此有可能是肿瘤免疫治疗的潜在靶点。
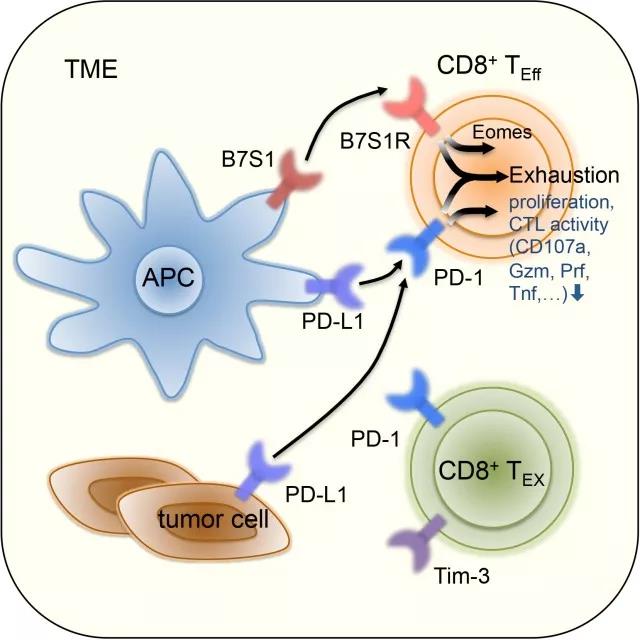
B7S1和pD-1信号共同促进T细胞衰竭
来自清华大学医学院的董晨课题组首先在肝细胞癌(HCC)病人的临床标本中检测B7S1及其受体的表达。在肿瘤微环境中,B7S1高表达于抗原呈递细胞(ApC),而它的潜在受体高表达于抗肿瘤效应淋巴细胞,pD-L1则主要表达于肿瘤细胞上,而且HCC病人肿瘤中CD8+ T细胞的功能缺陷与B7S1(而非pD-L1)的表达水平相关。
接着,他们在小鼠肿瘤模型中进一步验证B7S1在抗肿瘤免疫反应中的功能。他们发现阻断B7S1信号通路(B7S1敲除或anti-B7S1治疗)可抑制小鼠HCC (Hepa1-6)和淋巴瘤(A20、E.G7)的进展,在小鼠肿瘤中B7S1的受体(B7S1R)主要表达于CD8+ T细胞,而B7S1信号主要通过抑制ApC和CD8+ T细胞的相互作用来实现免疫抑制。然后,他们利用E.G7小鼠模型来研究B7S1信号产生免疫抑制的机制。首先,他们分析了肿瘤中表达B7S1R的CD8+ T细胞的功能状态。B7S1R随着CD8+ T细胞向肿瘤浸润或扩增而上调,在第12天达到最高点后下降,并与pD-1(而非Tim-3)共表达。体内表型分析和RNA-seq分析表明,pD-1+B7S1R+Tim-3-CD8+T细胞处于激活状态,而pD-1+B7S1R-Tim-3+CD8+T细胞处于衰竭状态。
通过对比野生型小鼠和B7S1敲除小鼠的表型,他们发现阻断B7S1信号通路可显著提高肿瘤浸润的CD8+ T细胞的数量和效应功能,并抑制T细胞衰竭的进程。体内外功能性实验表明,与T细胞衰竭密切相关的转录因子Eomes直接参与B7S1信号介导的免疫抑制。
由于B7S1R和pD-1共表达于肿瘤微环境中的CD8+ T细胞,而且阻断B7S1信号会导致pD-1的补偿性上调,董晨课题组猜测B7S1和pD-1信号可能会共同促进T细胞衰竭,同时阻断这两个通路可能会进一步增强抗肿瘤免疫力。于是他们对比了anti-B7S1、anti-pD-1单独用药和联合用药在E.G7和Hepa1-6小鼠肿瘤模型中的抑癌效果,研究结果表明,anti-B7S1和anti-pD-1联合治疗可更显著地抑制肿瘤生长。为研究联合治疗产生协同效应的机制,他们对比了经对照抗体、anti-B7S1、anti-pD-1和anti-B7S1+anti-pD-1治疗的小鼠中肿瘤抗原特异性CD8+ TIL的染色质开放区域(ATAC-seq)和转录组(RNA-seq)。CD8+ T细胞内大部分表观遗传修饰、基因表达以及信号通路由B7S1R和pD-1共同调控,表达于同一种细胞类型的B7S1R和pD-1可能通过作用于共同的下游信号分子来协同地抑制抗肿瘤CD8+ T细胞的功能,为anti-pD-1免疫治疗在癌症病人中应答率低的现象提供了理论依据。anti-B7S1和anti-pD-1治疗可进一步提高CD8+ TIL的效应功能和增殖存活能力,因此,在临床应用中,anti-B7S1和anti-pD-1联合用药有望进一步改善目前anti-pD-1免疫治疗的疗效。
另外一项研究中,董晨课题组发现 TRIM28缺失的T细胞几乎不能分化成Th17细胞。通过进一步的体内、体外实验,以及在Th17细胞中特异性敲除TRIM28后发现,TRIM28能够内源性地促进Th17细胞的分化,敲除该蛋白后T细胞的 IL-17表达下降、小鼠的自身性免疫疾病减轻。
此外,董晨课题组通过近一步在基因组水平的研究发现,TRIM28并不影响Th17细胞关键转录因子的表达,而是调节靶基因的组蛋白修饰、超级增强子形成以及染色质折叠。有趣的是,TRIM28在初始T细胞中就高表达,它在Il17/Il17f位点的结合特异地受到IL-6/STAT3信号的调控,从而引起靶基因的表观活化、RORγt的招募和功能发挥。该研究提出了外界的细胞因子信号影响Th17细胞表观活化的新机制,同时近一步阐释了STAT3和RORγt的层级关系,并且为TRIM28也能作为共活化分子提供了证据。
原文标题:
Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8+ T Cells
Epigenetic activation during T helper 17 cell differentiation is mediated by Tripartite motif containing 28































