利用参考样本对单细胞RNA测序技术进行多中心研究
12月21日《Nature Biotechnology》发表了一篇文章,比较了不同技术和不同实验室产生的不同单细胞RNA测序(scRNA-seq)数据集。Loma Linda大学的研究人员提出了在选择算法时需要指导的问题,从而能够对不同平台获得的不同数据类型进行精确的生物学解释。
研究人员使用两个特征良好的细胞参考样本(乳腺癌细胞和B细胞),分别或混合采集,比较不同scRNA序列平台和多个中心的预处理、标准化和批效应校正方法。虽然预处理和标准化有助于基因检测和细胞分类的可变性,但批量效应校正是迄今为止正确分类细胞的最重要因素。此外,scRNA-seq数据集的特性(例如,样本和细胞异质性以及所使用的平台)对于确定最佳的生物信息学方法至关重要。然而,当应用适当的生物信息学方法时,跨中心和平台的重复性很高。
这些发现为设计scRNA序列研究时优化平台和软件选择提供了实际指导。
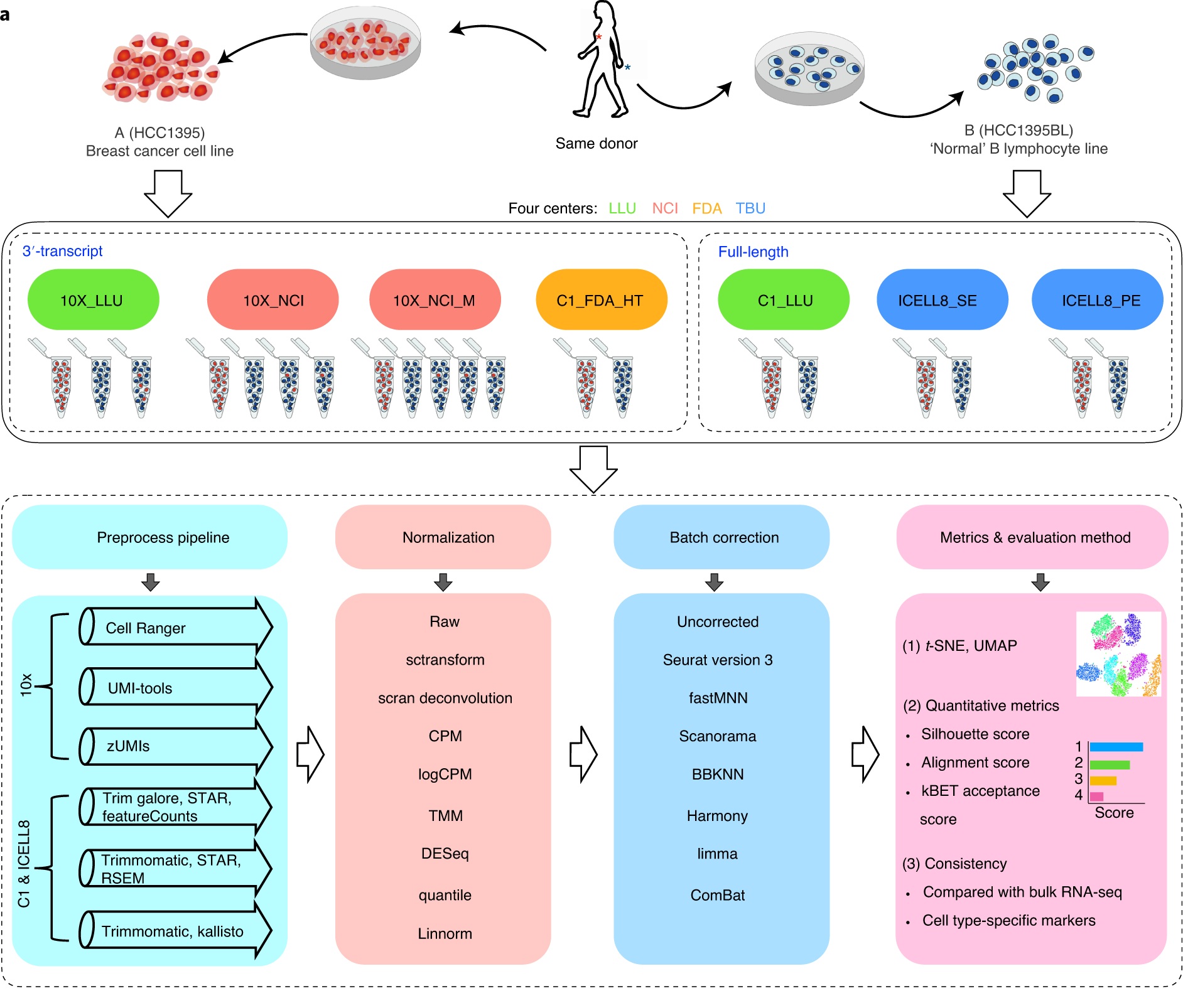
研究设计概要
使用两个参考细胞系(样本A,HCC1395;样本B,HCC1395BL)在四个平台(10x Genomics、FluidGM C1 HT、FluidGM C1和Takara Bio ICELL8)和四个检测点(LLU、NCI、FDA和TBU)生成scRNA序列数据。在LLU和NCI位点(10x),将10%或5%的癌细胞加入B淋巴细胞,制备混合单细胞捕获和文库构建。在NCI位点,用两种甲醇固定的细胞混合物(5%的癌细胞加入B淋巴细胞,名为固定细胞1和固定细胞2)进行单细胞捕获和文库构建。一组来自NCI的10x scRNA文库也使用一种较短的改良测序方法进行测序。BK-RNA-seq数据也从这些细胞系中获得,每个细胞系一式三份。
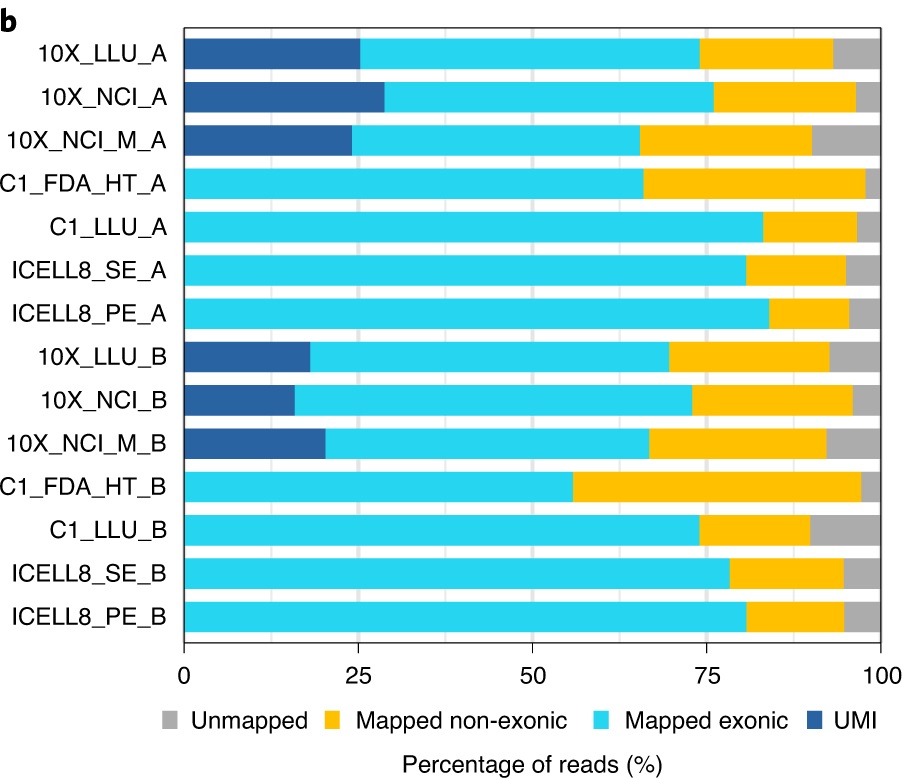
对于横跨14对数据集的乳腺癌细胞系(样本A)和B淋巴细胞系(样本B),显示了映射到外显子区域(蓝色)或非外显子区域(橙色)或未映射到人类基因组(灰色)的读数百分比。对于UMI方法(10x),深蓝色表示使用UMIs的外显子读取。
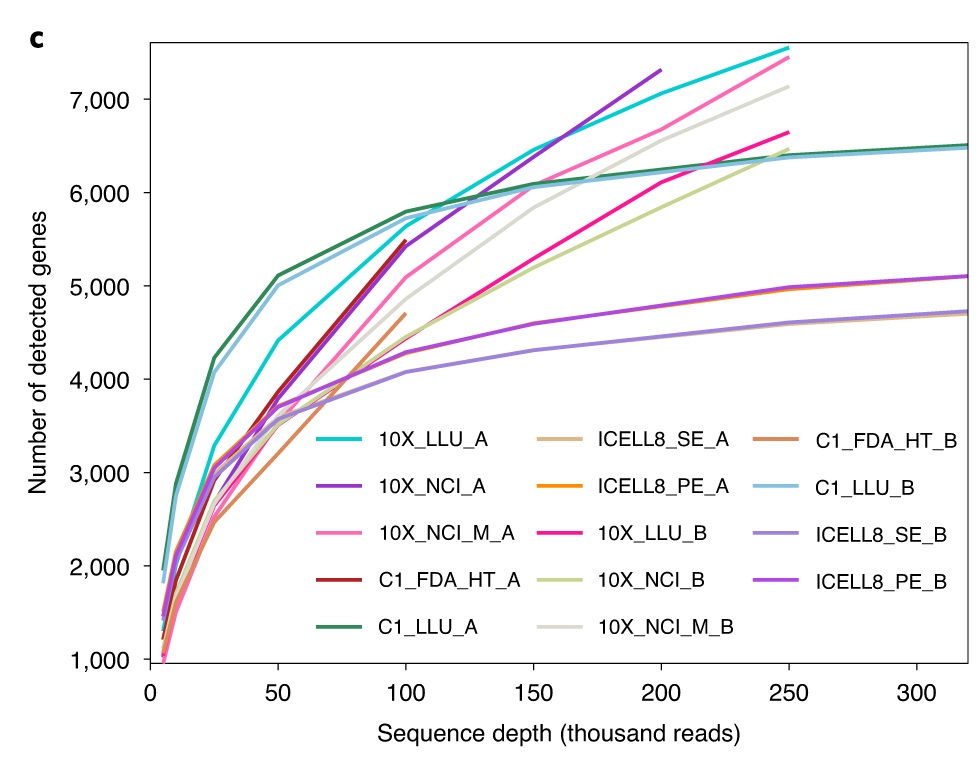
每个细胞在不同的测序读取深度检测到的基因的中位数。
原文检索:Chen W, Zhao Y, Chen X et al. (2020) A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples. Nat Biotechnol
(生物通:伍松)































