KDR基因变异与这种常见的先天性心脏病有关
法洛四联症(TOF)是最常见的紫绀型先天性心脏病,通常被认为是遗传和环境因素相互作用的结果。患者直系亲属和后代的先天性心脏病风险增加,说明了遗传因素的贡献。然而,对大多数患者而言,遗传因素仍然难以捉摸。
人们之前发现,血管内皮生长因子(VEGF)信号传导的失调与法洛四联症的发病机制有关,并发现一些患者编码血管内皮生长因子受体2的基因(KDR)存在罕见变异。不过,他们并不清楚KDR变异在法洛四联症发病机制中的作用。
荷兰阿姆斯特丹大学医学中心领导的一个国际研究团队近日在《Genetics in Medicine》杂志上发表文章,报道了KDR基因变异在法洛四联症发病中的作用。他们发现,罕见的KDR变异,特别是蛋白截短变异,与法洛四联症密切相关。未来在临床诊断中可以考虑对法洛四联症患者进行KDR筛查。
家族性TOF病例携带KDR变异
在这项研究中,研究人员首先对一个摩洛哥血统的家族进行了外显子组测序,该家族有两个孩子患有严重的法洛四联症(图1)。测序结果显示两名患者携带85个罕见变异。由于优先考虑双等位基因变异,故他们的焦点放在KDR杂合变异上:KDR-p.(Gly345Trp)和KDR-p.(Gly537Arg)。另一个未患病的姐妹没有携带任何KDR变异。
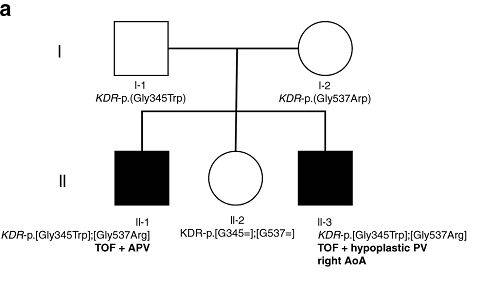
图1. 家族谱系
KDR基因编码了血管内皮生长因子受体2(VEGFR2)。此次发现的这两个变异位于VEGFR2的胞外免疫球蛋白样结构域4和5中,并且受影响的残基在进化上是保守的。此外,他们还在其他生物样本库中发现了带有罕见KDR变异的法洛四联症患者。
之后,研究人员委托赛业生物利用CRISpR-Cas9靶向系统生成了基因敲入小鼠品系,每个品系分别携带了两个KDR变异的小鼠同源物。他们发现,Kdr-p.(Gly535Arg)(与KDR-p.(Gly537Arg)同源)纯合敲入小鼠再现了Kdr缺失小鼠的表型,表明这种纯合变异对发育有严重的影响。
KDR变异导致VEGFR2磷酸化降低
为了阐明两个KDR变异的生物学机制,研究人员随后分析了VEGFR2上酪氨酸1175(Tyr1175)位点的磷酸化。这是VEGFR2的主要磷酸化位点之一,对下游信号级联中蛋白质的募集至关重要。
他们用VEGF165来刺激异源表达两个变异或野生型KDR(对照)的HEK 293T细胞。Western blot分析结果表明,与野生型相比,这两个变异导致Tyr1175位点的磷酸化水平显著降低。同样地,对于来自敲入小鼠胚胎的卵黄囊细胞,VEGFR2的磷酸化水平也明显降低(图2)。其中,KDR-p.(Gly537Arg)变异的功能影响更为严重。
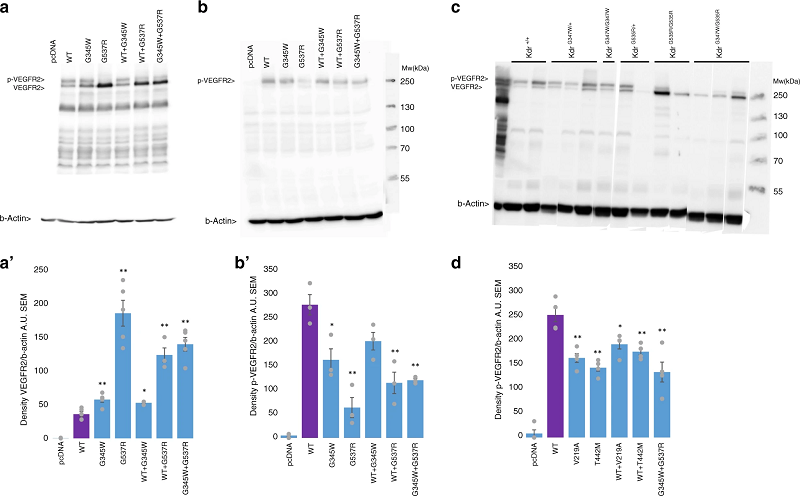
图2. 磷酸化分析
最后,研究人员通过比较罕见变异在法洛四联症患者和对照上的负荷来探索KDR变异的致病作用。他们对一组共1,569名欧洲血统的法洛四联症患者开展变异负荷分析。与对照组相比,法洛四联症患者出现蛋白质截短变异(pTV)的比例大约高了46倍。1,569个病例中有9个pTV,而56,699个对照中有7个pTV(p = 7 × 10-11)。同时,法洛四联症患者的KDR错义变异比例也更高,但没有统计学意义。
结论
作者认为,罕见的KDR变异,特别是蛋白质截短变异,与法洛四联症密切相关,可能存在不同的遗传模式。有了遗传学以及体内外功能分析的支持,他们认为VEGFR2的功能缺失是引起法洛四联症的机制之一,未来在临床诊断中可考虑对法洛四联症患者进行KDR筛查。
原文检索
Škorić-Milosavljević, D., Lahrouchi, N., Bosada, F.M. et al. Rare variants in KDR, encoding VEGF Receptor 2, are associated with tetralogy of Fallot. Genet Med (2021). https://doi.org/10.1038/s41436-021-01212-y































